Food and Drug Administration (FDA) regulations have become the new pathway to riches for the pharmaceutical industry.
First, there was generic colchicine, used for years and years to treat gout for pennies a pill. The only problem was, there wasn't an FDA trial proving colchicine's efficacy in the treatment of gout. Takeda Pharmaceutical, seeing the opening, performed a trial and rebranded the formerly generic colchicine to Colcrys®, "the only authorized generic indicated to prevent and treat gout attacks." And how much does Colcrys® cost? Just $203 for thirty tablets at Costco.
But that's not all.
Today I learned that generic vasopressin (which can be stored at room temperature in stable form on crash carts), must be switched to the FDA-approved brand called Vasostrict® that requires dilution and refrigeration. It seems the generic form of vasopressin will no longer be available to be kept on crash carts since it's not "FDA-approved" for the indication of "increasing blood pressure in adults with vasodilatory shock (post-cardiotomy or sepsis) who remain hypotensive despite fluids and catecholamines." Vasostrict®, on the other hand, is "now the first and only vasopressin injection, USP, product with an NDA approved by the FDA." The catch is, it must diluted before use and discarded after 18 hrs (or after 24 hrs if refrigerated). This little regulatory quirk is a big deal for America's hospitals looking to save costs.
But hey, why should we worry about costs in health care? After all, you can never be too safe.
-Wes
Showing posts with label FDA. Show all posts
Showing posts with label FDA. Show all posts
Tuesday, January 20, 2015
Monday, November 17, 2014
J. Rod Gimbel: Crowdsourcing a Consumer Safety Issue
The following is a guest post by J. Rod Gimbel, MD, a cardiac electrophysiologist from Knoxville, TN who has written extensively on the issue of electronic surveillance systems and electromagnetic interference with cardiac implantable electronic devices:
-Wes
I’d like to express my appreciation for allowing me to guest post in this space.This is an important effort that Rod is undertaking on behalf of patients with CIEDs. I hope patients and health care providers will come forward with examples of EAS systems or EAS interference in their locales to assist him in this important consumer safety effort.
This is about crowdsourcing a consumer safety issue; specifically the public safety of consumers who happen to have CIEDs (cardiac implantable electronic devices) such as pacemakers or implantable defibrillators (ICD). Nearly 2 million such consumers (patients) have CIEDs in the U.S. alone. As you know, these devices are susceptible to EMI (electromagnetic interference). Simply put, the lead(s) act like antennas and can pick up stray EMI from any number of sources and cause the device to malfunction by either withholding therapy (no pacing or ICD rescue therapy) or through delivery of inappropriate therapy (delivering pacing output or shocks where none is needed). Either situation can be life threatening.
One source of EMI that can affect a CIED patient is electronic article surveillance system (EAS). Such systems are widely used by retailers (ref) to deter and prevent store theft, a problem commonly referred to as “shrinkage”.
About 8 years ago, an ICD patient that I was caring for received inappropriate shocks from his ICD after being near an EAS system located in a big box retailer. A colleague of mine related a similar situation where a pacemaker dependent patient reported syncope in the proximity of an EAS system after her pacemaker inhibited in response to the EMI from the EAS system. These were two disturbing, potentially life threatening events. In hopes of raising awareness of this serious problem (EAS-CIED interaction), we generated a manuscript detailing the events that was published in 2007 in the Mayo Clinic Proceedings. Notably, the New York Times picked up the story. Others have published similar unfortunate misadventures between patients and EAS systems.
Several common sense recommendations have been made in this area; recommendations that preserve a retailer’s right to deter and reduce theft (a legitimate concern), but still protect CIED patients from adverse interactions with EAS systems. For instance, after receiving reports of several adverse events caused by EAS systems the Food and Drug Administration (FDA) issued a “Safety Communication” and noted:
Beyond this, we and others also suggested:
- Be aware that EAS systems may be hidden/camouflaged in entrances and exits where they are not readily visible in many commercial establishments.
- Do not stay near the EAS system or metal detector longer than is necessary and do not lean against the system.
- Retailers should not "camouflage" the EAS pedestals with advertising as this may prevent customers with devices from recognizing the threat and may actually draw device patients toward the EAS pedestal.
- Retailers should not place goods and services near the EAS systems that effectively encourage the patients to violate the "don't linger, don't lean" dictum that physicians tell patients who have devices.
It seems entirely reasonable to suggest a shared responsibility between medical device professionals, device patients, retailers, and EAS system manufacturers. It was hoped EAS manufactures and retailers would do their part and embrace these simple recommendations and help make retail spaces safe for those with implantable devices. Unfortunately, this does not seem to always be the case.
Figure 1: Bench to "relax" placed adjacent to a camouflaged EAS pedestal at big box retailer.
Figure 2: Chair to "relax" placed adjacent to EAS pedestal while patient waits for prescription to be filled at retail pharmacy store. These pictures were taken in the last several months around the country. Clearly, the juxtaposition of EAS systems and consumer areas may undermine the dictum “don’t linger, don’t lean” and leave device patients in harm’s way. Who then, is responsible for the safety of device patients in this situation?
Figure 3: Complimentary coffee station where device patients might linger placed adjacent to EAS pedestal at big box retailer
Finally, perhaps in an attempt to thwart a determined and “informed shoplifter” who may employ several methods that might undermine the effectiveness of EAS systems, “there are also concerns that some installations are purposefully configured to exceed the rated specifications of the manufacturer, thereby exceeding tested and certified magnetic field levels.” This may increase further the risk of adverse reactions experienced by device patients when near EAS systems.
Now for the crowdsourcing part:
A presentation on this topic (CIED-EAS interactions) to an extra governmental regulatory group helping set standards for the device industry is to be given soon. This presentation will be to a number of interested parties including representatives of the EAS manufactures, device manufactures, and the FDA. As noted above, the “event rate” of these interactions is rather low, but as has been suggested significant under-reporting may obscure the true significance of the problem. It is surely recognize that not everyone has the time or inclination to write up adverse events for publication or inclusion in a database. Perhaps, some events go entirely unrecognized for what they really are, being passed off as “Oh, Mom passed out at the store today, but she’s OK now”.
With your help a strong presentation and case can be made emphasizing CIED-EAS interactions are an important public safety issue. Your voice and concerns can be heard. First off, send pictures where you see EAS systems placed in a manner that might endanger a device patient (like the ones shown above). Cell phone pictures are just fine. Second, if you are a health care provider or patient, please send any “events” that you may have experienced describing an adverse interaction between an EAS system and pacemakers and or ICDs. Please post the items here or send items of interest to J. Rod Gimbel, MD (gimbeljr@gmail.com). Your response is of course appreciated and in confidence and any presentation of the material provided will be anonymized. Upon completion of the presentation, a link will be posted here.



-Wes
Wednesday, February 19, 2014
Are Drug Letters Harming Our Patients?
 |
| One week's worth of drug letters |
Here's a typical (paraphrased) letter that has me most concerned:
"Dear Mr or Ms Patient:Note that all novel oral anticoagulant medications carry a very specific black-box warning from the FDA regarding discontinuation of these medications. Here's the black box warning for rivaroxaban (Xarelto):
This letter is to inform you that Insurance Plan XYZ ("the plan") has provided you with a temporary supply of the following prescription:
(Insert your favorite novel oral anticoagulant name/dose here)
As described in more detail on the following page, this drug is either not included on our covered drug list (formulary) or included on the drug list, but has certain limits on it.
Because you are within the first 90 days of coverage this plan year, TooBig Insurance Company is required to provide you with at least a 31-day supply of this drug. The supply is less if the prescription is written for less and does not have refills...
It is important you understand this is a temporary supply of this drug. Before this supply ends, you should talk with your doctor to decide if you should change your prescription. (Emphasis mine) If your doctor feels that the coverage options listed on the following page are not clinically appropriate for your situation, your doctor can request a formulary exception from TooBig Insurance Company to continue coverage of this drug.
If you need help requesting an exception, please call TooBig Insurance Company Customer Service listed on the back of your member ID card."
"A. PREMATURE DISCONTINUATION OF XARELTO® INCREASES THE RISK OF THROMBOTIC EVENTSShould insurance companies be allowed to restrict the dispensing of any medication where doing so puts patients at risk of injury? Why are insurance companies allowed to potentially violate FDA-mandated black box warnings if they do so? How many lives are being placed at risk by this practice? What about the patients that are blind or can't read? What happens to them?
Premature discontinuation of any oral anticoagulant, including XARELTO®, increases the risk of thrombotic events. If anticoagulation with XARELTO® is discontinued for a reason other than pathological bleeding or completion of a course of therapy, consider coverage with another anticoagulant."
On a more selfish note: why must the physician suffer the liability consequences of an insurer's profit-driven actions that disregards the safety and well-being of our patients?
Why?
-Wes
Friday, July 26, 2013
An Open Letter to Patient's With Pre-excited Afib and Ischemic VT
Dear Mr. or Ms. Patient With Pre-excited Afib or Ischemic VT:
I just wanted to let you know, if you come to our ER, you are screwed. Currently, our best drug to deal with your arrhythmias of pre-excited atrial fibrillation (afib) or ischemic ventricular tachycardia is not available anywhere: procainamide. It seems the one drug company who makes this drug (Hospira) has a few manufacturing delays (oops), so the drug is on backorder.
So come ready to have your heart shocked.
Hopefully we'll have some analgesic or anesthetic drugs available in our pharmacy that aren't on backorder so you won't feel your cardioversion.
Wishing you the best, as always...
-Wes
I just wanted to let you know, if you come to our ER, you are screwed. Currently, our best drug to deal with your arrhythmias of pre-excited atrial fibrillation (afib) or ischemic ventricular tachycardia is not available anywhere: procainamide. It seems the one drug company who makes this drug (Hospira) has a few manufacturing delays (oops), so the drug is on backorder.
So come ready to have your heart shocked.
Hopefully we'll have some analgesic or anesthetic drugs available in our pharmacy that aren't on backorder so you won't feel your cardioversion.
Wishing you the best, as always...
-Wes
Thursday, March 14, 2013
The Generality of the FDA's Recommendations on Zithromax
Tuesday's warning about Zithromax causing heart irregularities reportedly came after the FDA completed its own review of a New England Journal of Medicine article published in May 2012 and after reviewing additional data provided by Pfizer. I have already voiced my concerns over the way this study was conducted in the first place since the data upon which the authors' risk estimates were made were shoddy at best. Still, it is quite obvious to those of us in the business of cardiac arrhythmias that Zithromax, when combined with other medications likely to prolong QT interval or given to sick patients, could increase one's arrhythmic risk.
But we should look closer at the FDA's new Drug Safety Communication on Zithromax which says: "Health care professionals should consider the risk of torsades de pointes and fatal heart rhythms with azithromycin when considering treatment options for patients who are already at risk for cardiovascular events."
Gee, who knew?
If we probe deeper, we find the FDA recommends caution in prescribing antibiotics in patients likely to develop life-threatening cardiac arrhythmias like those with advanced age who are hospitalized, have low potassium or magnesium blood levels, or have slow heart rhythms. The truth be known, each one of these conditions in and of themselves increases the risk of QT prolongation and makes them more susceptable to cardiac arrhythmias irrespective of the drug admininstered.
So how helpful was the FDA's warning on Zithromax to America's doctors?
Not very.
Perhaps we should add that warning to every drug out there, eh?
-Wes
But we should look closer at the FDA's new Drug Safety Communication on Zithromax which says: "Health care professionals should consider the risk of torsades de pointes and fatal heart rhythms with azithromycin when considering treatment options for patients who are already at risk for cardiovascular events."
Gee, who knew?
If we probe deeper, we find the FDA recommends caution in prescribing antibiotics in patients likely to develop life-threatening cardiac arrhythmias like those with advanced age who are hospitalized, have low potassium or magnesium blood levels, or have slow heart rhythms. The truth be known, each one of these conditions in and of themselves increases the risk of QT prolongation and makes them more susceptable to cardiac arrhythmias irrespective of the drug admininstered.
So how helpful was the FDA's warning on Zithromax to America's doctors?
Not very.
Perhaps we should add that warning to every drug out there, eh?
-Wes
Tuesday, November 27, 2012
Subcutaneous ICD: A Lesson in FDA Approval
This morning, a nice article in the Boston Globe appeared on Boston Scientific's subcutaneous implantable cardiac defibrillator (S-ICD) that recently received FDA approval. It was a fairly balanced article, one that touched ever-so-briefly on the pros and cons of a subcutaneous device to treat life-threatening cardiac arrhythmias using a medical device that does not require an internal wire inside the heart, but rather a sensing and shocking lead that is tunneled accross the chest under the skin (see my prior post on the details here).
But what I found most interesting in the article was the patient who was recommended for the device: a dialysis patient, was a patient who was specifically excluded from the FDA trials to approve the device (specifically, patients with GFR < 29 were excluded).
If you are a company that wants to get a device approved by the FDA, you want the best chances of having the fewest complications possible with new gadgets in medicine. Because sick dialysis patients have a way of having more complications with device implants, and the device companies know this, they are not included in trials to get a device approved.
Those of us who deal with ICDs in dialysis patients recognize the problems when ICD leads and dialysis catheters co-exist in the same vascular tree: the odds of infecting the ICD lead system is extraordinarily high. In fact, the overall mortality advantage of ICDs in dialysis patients is much less than patients not on dialysis. For this clinical circumstance, subcutaneous ICDs would seem to clearly be the better choice.
But dialysis patients are beset by another problem: challenges with potassium level regulation. Periods of hyperkalemia are quite common in dialysis patients and hyperkalemia commonly causes severe bradycardia. In these patients, pacing could maintain a patient's heart rate until their dialysis could be adjusted to lower their potassium and thereby improve their cardiac function. Subcutaneous ICDs do not have pacing capabilities, however.
So the reality of the effectiveness of the subcutaneous ICD to prolong life is uncertain in dialysis patients. This theory has never been tested - we just tend to think it makes intuitive sense to apply a new technology when other medical device choices are not perfect either.
The subcutaneous ICD FDA approval process is a good example of how clinical dogma spreads amongst doctors and patients without any proof of a device's effectiveness in some subselected patient populations. A new technology is approved by the FDA and receives a governmental stamp of approval. Doctors, then, serve as well-meaning spokespersons. The media gushes over the latest and greatest technology. And sales explode...
... all while some patients in whom the device is deployed doesn't realize the safety and efficancy of that device was never tested in their particular clinical circumstance.
We should remember that exclusion criteria in FDA clinical trials are important. They tell us who might and who might not benefit from a new technology. Too often we forget this.
And so does the FDA.
-Wes
But what I found most interesting in the article was the patient who was recommended for the device: a dialysis patient, was a patient who was specifically excluded from the FDA trials to approve the device (specifically, patients with GFR < 29 were excluded).
If you are a company that wants to get a device approved by the FDA, you want the best chances of having the fewest complications possible with new gadgets in medicine. Because sick dialysis patients have a way of having more complications with device implants, and the device companies know this, they are not included in trials to get a device approved.
Those of us who deal with ICDs in dialysis patients recognize the problems when ICD leads and dialysis catheters co-exist in the same vascular tree: the odds of infecting the ICD lead system is extraordinarily high. In fact, the overall mortality advantage of ICDs in dialysis patients is much less than patients not on dialysis. For this clinical circumstance, subcutaneous ICDs would seem to clearly be the better choice.
But dialysis patients are beset by another problem: challenges with potassium level regulation. Periods of hyperkalemia are quite common in dialysis patients and hyperkalemia commonly causes severe bradycardia. In these patients, pacing could maintain a patient's heart rate until their dialysis could be adjusted to lower their potassium and thereby improve their cardiac function. Subcutaneous ICDs do not have pacing capabilities, however.
So the reality of the effectiveness of the subcutaneous ICD to prolong life is uncertain in dialysis patients. This theory has never been tested - we just tend to think it makes intuitive sense to apply a new technology when other medical device choices are not perfect either.
The subcutaneous ICD FDA approval process is a good example of how clinical dogma spreads amongst doctors and patients without any proof of a device's effectiveness in some subselected patient populations. A new technology is approved by the FDA and receives a governmental stamp of approval. Doctors, then, serve as well-meaning spokespersons. The media gushes over the latest and greatest technology. And sales explode...
... all while some patients in whom the device is deployed doesn't realize the safety and efficancy of that device was never tested in their particular clinical circumstance.
We should remember that exclusion criteria in FDA clinical trials are important. They tell us who might and who might not benefit from a new technology. Too often we forget this.
And so does the FDA.
-Wes
Thursday, July 26, 2012
How the iPhone Might Disrupt The Medical Device Industry
Doctors wanting to determine a patient's atrial fibrillation burden have a myriad of technologies at their disposal: 24-hour Holter monitors, 30-day event monitors that are triggered by an abnormal heart rhythm or by the patient themselves, a 7-14 day patch monitor that records every heart beat and is later processed offlineto quanitate the arrhythmia, or perhaps an surgically-implanted event recorder that automatically stores extremes of heart rate or the surface ECG when symptoms are felt by the patient. The cost of these devices ranges from the hundreds to thousands of dollars to use.
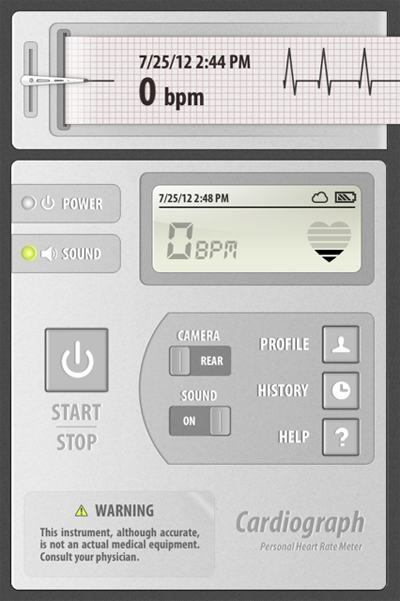
Today in my clinic, a patient brought me her atrial fibrillation burden history on her iPhone and it cost her less than a $10 co-pay. For $1.99 US, she downloaded the iPhone app Cardiograph to her iPhone.

Every time she feels a symptom, she places her index finder over the camera on the phone, waits a bit, and records a make-believe rhythm strip representing each heart rhythm. With it, comes the date and time. When the rhythm is in sinus, she learned that her heart rhythm was typically in the 60's at rest:

When the rhythm was in afib, it was considerably higher and sometimes displayed an irregular rhythm:

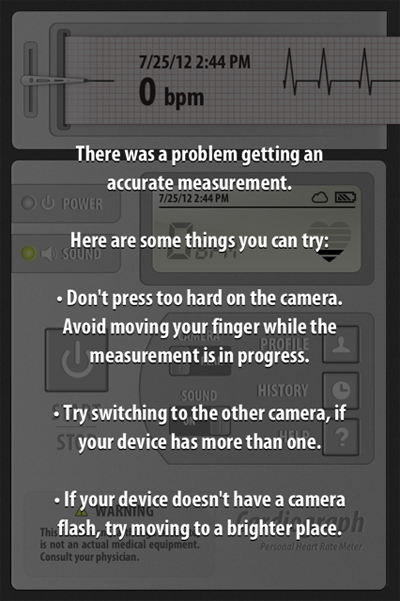
or sometimes it displayed an error message:

I got a relative picture of how often she was having afib and she got the opportunity to help me with her care.
Was this a medical device? No, it was an iPhone app.
Was it perfect? No it wasn't. I certainly couldn't differentiate frequent PAC's or PVC's from atrial fibrillation reliably. It was NOT an EKG after all. But we were past that point in her evaluation. I just needed to know how often she was having her known paroxysmal atrial fibrillation and she wanted to keep a convenient record of her episodes.
Was it helpful in this case? Absolutely.
More importantly, she just saved herself and the health care system a ton of money.
Welcome, my friends, to the era of patient-empowered, individualized medicine and a whole new era of patient care. Now, if we can just keep the FDA from screwing things up.
-Wes
PS: I have no commercial interest in the Cardiograph app and do not endorse it as a standard of care, but merely use this case to demonstrate how innovation can facilitate cheaper, equally-effective health care in some cases. I'd also like to thank my patient for allowing me to use her screen shots.
Today in my clinic, a patient brought me her atrial fibrillation burden history on her iPhone and it cost her less than a $10 co-pay. For $1.99 US, she downloaded the iPhone app Cardiograph to her iPhone.
Every time she feels a symptom, she places her index finder over the camera on the phone, waits a bit, and records a make-believe rhythm strip representing each heart rhythm. With it, comes the date and time. When the rhythm is in sinus, she learned that her heart rhythm was typically in the 60's at rest:

When the rhythm was in afib, it was considerably higher and sometimes displayed an irregular rhythm:
or sometimes it displayed an error message:
I got a relative picture of how often she was having afib and she got the opportunity to help me with her care.
Was this a medical device? No, it was an iPhone app.
Was it perfect? No it wasn't. I certainly couldn't differentiate frequent PAC's or PVC's from atrial fibrillation reliably. It was NOT an EKG after all. But we were past that point in her evaluation. I just needed to know how often she was having her known paroxysmal atrial fibrillation and she wanted to keep a convenient record of her episodes.
Was it helpful in this case? Absolutely.
More importantly, she just saved herself and the health care system a ton of money.
Welcome, my friends, to the era of patient-empowered, individualized medicine and a whole new era of patient care. Now, if we can just keep the FDA from screwing things up.
-Wes
PS: I have no commercial interest in the Cardiograph app and do not endorse it as a standard of care, but merely use this case to demonstrate how innovation can facilitate cheaper, equally-effective health care in some cases. I'd also like to thank my patient for allowing me to use her screen shots.
Saturday, September 17, 2011
The FDA Drug Safety Communication I'd Like to See
FDA (Food and Drug Administration): Drug Safety Communication - Excessive FDA Drug Safety Communications
[Posted 09/17/2011]
AUDIENCE: Physicians, Patients
ISSUE: FDA has notified healthcare professionals and patients of ongoing safety review and labeling changes for drugs such as Zofran (ondansetron, ondansetron hydrochloride and generics). It has been determined by the FDA that the FDA may be on the brink of creating a "Boy Who Cried Wolf" scenario by notifying doctors of prolonged QT intervals in drugs twenty years after their first market approval. Further, notifications such as these included two studies from 2005 and 2008 which were both written by the same lead author. The FDA realizes it is now 2011. An FDA investigation to determine if this same author now works for the FDA is ongoing.
BACKGROUND: Zofran (ondansetron) is in a class of medications called 5-HT3 receptor antagonists and is one of the few effective and remarklably safe drugs that doctors have used for years. While QT-prolonging effects with this medication have been seen in small controlled trials (see the FDA's references), realize that limitations to at least one of those trials existed, since 21% of an 85-patient study had prolonged QT before the drug was administered. Also note that the second trial only included a whopping 8 patients in the study group.
RECOMMENDATION: The FDA recommends that FDA Drug Safety Communications be inspected closely. Errant recommendations based on dated, single-center studies are becoming the norm and doctors are advised that excessive FDA Safety Communications may be forthcoming because certain government exployees are working hard to perform exhaustive literature searches with litle relevance to clinical experience.
That is all.
As always, healthcare professionals and patients are encouraged to report adverse events or side effects related to the use of these products to the FDA's MedWatch Safety Information and Adverse Event Reporting Program:
Complete and submit the report Online: www.fda.gov/MedWatch/report.htm
Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
[09/17/2011 - Drug Safety Communication - FDA]
-Wes
See also WhiteCoat's take.
Thursday, September 15, 2011
We've Hit the Jackpot With A Million Hearts
I don't know what I was thinking with my last post about the Health and Human Services' Million Hearts initiative. I thought the whole point of this program was to save money. At the time, I was less than optimistic that the government could acurately reach their goal given the problems with many of the principles behind their program. For instance, maybe it was just me, but how typing on an electronic medical record system would save those lives was lost on me.
But at the time, I had no idea this whole campaign was based on fear.
Watch this introductory video I found on the brand new Million Hearts website, all paid for (of course) with your tax payer dollars:
Listen to all that horrible stuff that can happen to you! And they look like such nice, ordinary people. Man, if that doesn't scare you into seeing your doctor, I don't know what will!
I have now seen the light: this is going to be awesome! (* Cha-ching! *)
You know: terrified, low risk people clamoring for an appointment insisting on all kinds of blood draws, testing, EKGs, and sophisticated imaging tests! And for all those tests with false positives, maybe an angio, too! We're about to make a fortune on this program all in the name of "cost savings!" Baby, there's NOTHING like screening low risk people when it comes to making money: Yeeeeeee haaaaaa!
Boys and girls, step right up! Help our heath care system save money, will you? (* Yuck yuck yuck *) After all (wink wink) it's about saving a million lives! You did see that video above, didn't you? No? Make sure you do: this is really BAD stuff!
Boy, no WONDER the American Medical Association, the American Heart Association, the American College of Cardiology, Walgreens, United Healthcare and a whole host of other "private interests" want to support this program: it's not just about the hearts, it's about the Benjamins!
Even better: they're on Facebook and Twitter! (Hope their video goes viral! Pssst: here's the link: http://www.youtube.com/embed/poc1eTuJJfc)
Absolutely, positively, brilliant. (I hope our health care system's marketing team is watching.)
After all, nothing sells like fear.
-Wes
But at the time, I had no idea this whole campaign was based on fear.
Watch this introductory video I found on the brand new Million Hearts website, all paid for (of course) with your tax payer dollars:
Listen to all that horrible stuff that can happen to you! And they look like such nice, ordinary people. Man, if that doesn't scare you into seeing your doctor, I don't know what will!
I have now seen the light: this is going to be awesome! (* Cha-ching! *)
You know: terrified, low risk people clamoring for an appointment insisting on all kinds of blood draws, testing, EKGs, and sophisticated imaging tests! And for all those tests with false positives, maybe an angio, too! We're about to make a fortune on this program all in the name of "cost savings!" Baby, there's NOTHING like screening low risk people when it comes to making money: Yeeeeeee haaaaaa!
Boys and girls, step right up! Help our heath care system save money, will you? (* Yuck yuck yuck *) After all (wink wink) it's about saving a million lives! You did see that video above, didn't you? No? Make sure you do: this is really BAD stuff!
Boy, no WONDER the American Medical Association, the American Heart Association, the American College of Cardiology, Walgreens, United Healthcare and a whole host of other "private interests" want to support this program: it's not just about the hearts, it's about the Benjamins!
Even better: they're on Facebook and Twitter! (Hope their video goes viral! Pssst: here's the link: http://www.youtube.com/embed/poc1eTuJJfc)
Absolutely, positively, brilliant. (I hope our health care system's marketing team is watching.)
After all, nothing sells like fear.
-Wes
Tuesday, September 13, 2011
For Your Safety: How to Prescribe an Antiarrhythmic Drug
First, read an 8-page treatment guideline.
Next, read the 26-page prescribing information.
Next, read the patient's 3-page medication guide.
Next, fill out a database form for the drug company to add yourself to their new-and-improved database (even though you've already registered with them once before) once again before January 1, 2012.
Next, (and this is important), agree to the following:
I have no idea where the every "three month" suggested lab testing and EKG requirement came from and have to wonder: if I sign their form, what is the economic burden to patients who must pay for this mandated testing? Who will enforce these requirements? Will following these recommendations absolve a doctor from potential liability with this medication? Do any of these requirements tell us anything about problems with the medication after they are prescribed?
No.
But these requirements make our regulators feel good while ignoring the very real financial and time costs that they impose directly on patients and doctors.
For instance, I called the company and asked if they would provide me their required Medication Guide to give my patients. They politely suggested I take the time to print out the guide from their TikosynREMS.com website for each patient. Or better yet, maybe I could have my staff make copies of the three-page form for my patients!
Gee, thanks.
No doubt we'll soon have PradaxaREMS.com, EliquisREMS.com, SeroquelREMS.com, ViagraREMS.com and hundreds to thousands of other REMS websites, each with their own prescribing guides, patient medication quides and database registrations.
Safety first, remember?
I just wonder when I'll have the time to see all of my other patients.
-Wes
Next, read the 26-page prescribing information.
Next, read the patient's 3-page medication guide.
Next, fill out a database form for the drug company to add yourself to their new-and-improved database (even though you've already registered with them once before) once again before January 1, 2012.
Next, (and this is important), agree to the following:
- That patients initiated or reinitiated on the medication should be admitted for three days and a bunch of other stuff with that,
- That you understand that following the treatment initiation and dosing guidelines in the drug's label will decrease the chance of a drug-induced arrhythmia
- That you will inform the patients of the risk of arrhythmias
- That you will need to see them for an EKG and blood tests every three months
- That you will provide a Medication Guide to each patient at the initiation and re-initiation of the drug's therapy and that you will review the contents of the medication guide personally with each patient.
I have no idea where the every "three month" suggested lab testing and EKG requirement came from and have to wonder: if I sign their form, what is the economic burden to patients who must pay for this mandated testing? Who will enforce these requirements? Will following these recommendations absolve a doctor from potential liability with this medication? Do any of these requirements tell us anything about problems with the medication after they are prescribed?
No.
But these requirements make our regulators feel good while ignoring the very real financial and time costs that they impose directly on patients and doctors.
For instance, I called the company and asked if they would provide me their required Medication Guide to give my patients. They politely suggested I take the time to print out the guide from their TikosynREMS.com website for each patient. Or better yet, maybe I could have my staff make copies of the three-page form for my patients!
Gee, thanks.
No doubt we'll soon have PradaxaREMS.com, EliquisREMS.com, SeroquelREMS.com, ViagraREMS.com and hundreds to thousands of other REMS websites, each with their own prescribing guides, patient medication quides and database registrations.
Safety first, remember?
I just wonder when I'll have the time to see all of my other patients.
-Wes
Monday, September 12, 2011
Elementary, My Dear Watson

We should ask ourselves that question now, especially since your insurer might be using Watson to mine your electronic medical record to determine if your claim will be paid.
Wellpoint, one of the nation's largest insurers is joining forces with IBM to use Watson as a medical decision support tool:
The first Watson deployment would come early next year with WellPoint nurses who manage complex patient cases and review treatment requests from medical providers.As such, shouldn't such a computer program be subject to the same rigors as the medical device industry to achieve FDA clearance? Would regulators even be able to comprehend how Watson reaches it's comclusions as it culls through "200 million pages of information in less than three seconds?"
Or will this device be grandfathered in under a 510K exemption? After all, it's just a computer right? Seriously, what could go wrong with clinical decisions based on cold, hard, incomplete data sets that are extrapolated to patients?
Wait, I know!
It's the Daily Double!
-Wes
Thursday, September 01, 2011
Of Pens and Payoffs
It's the ultimate irony to this casual observer.
Remember when doctors were chastised for accepting a pen worth pennies from a pharmaceutical company due to the pharmaceutical industry's pervasive marketing techniques that swayed the prescribing practices of millions of doctors?
Bad doctors.
We should have known better: all that covert marketing influence created by all those pens. No doubt thanks to that former practice we were single-handedly raising health care costs for Americans every time we looked at our pop-up Viagra pens. Man were we dogs!
Dirty. Rotten. Scoundrels. All.
So thank GOODNESS that the FDA can raise the fees it extracts from those same drug companies for their purposes! Certainly there would never be any influence on members of the FDA, especially since the negotiations between the FDA and the pharmaceutical lobbying organization were "relatively smooth." Let's see: A 6% increase to the 62% of the $930 million the FDA spends annually to review new pharmaceutical applications?
Around here, we call this "The Chicago Way:"
It's just a little money for a few pens.
Really."
-Wes
Remember when doctors were chastised for accepting a pen worth pennies from a pharmaceutical company due to the pharmaceutical industry's pervasive marketing techniques that swayed the prescribing practices of millions of doctors?
Bad doctors.
We should have known better: all that covert marketing influence created by all those pens. No doubt thanks to that former practice we were single-handedly raising health care costs for Americans every time we looked at our pop-up Viagra pens. Man were we dogs!
Dirty. Rotten. Scoundrels. All.
So thank GOODNESS that the FDA can raise the fees it extracts from those same drug companies for their purposes! Certainly there would never be any influence on members of the FDA, especially since the negotiations between the FDA and the pharmaceutical lobbying organization were "relatively smooth." Let's see: A 6% increase to the 62% of the $930 million the FDA spends annually to review new pharmaceutical applications?
Around here, we call this "The Chicago Way:"
They pull a knife, you pull a gun. He sends one of yours to the hospital, you send one of his to the morgue. That's the Chicago way."Influence? What influence? I'm not seein' no stinkin' influence of these payments to our government officials! No way!
–Jim Malone, "The Untouchables"
It's just a little money for a few pens.
Really."
-Wes
Wednesday, August 17, 2011
The Clinical Costs of Pharmachologic Post-Market Surveillance
Every drug a doctor prescribes requires an intimate knowledge of the drug's pharmacology, side effects, and possible drug interactions. Nowhere is this more true than antiarrhythic drugs. Concern over side effects with government regulators has reached a fever pitch since there is realization that all the pre-market randomized controlled trials often fail to identify later problems with medications. A classic example of this is dronedarone, initially heralded as a "safer" amiodarone substitute, but was later implicated in rare instances of fulminant hepatic failure.
While post-market surveillance of medications is both necessary and warranted, it is interesting to me how the grunt work of this surveillance (and most other grand regulatory schemes) falls squarely on the backs of physicians rather than the drug companies who manufacture and profit from the medications.
Case in point: the FDA's REMS program. REMS stands for "Risk Evaluation and Mitigation Strategy" and is a program developed by the FDA "to manage known or potential serious risks associated with a drug product. It is required by the Food and Drug Administration (FDA) to ensure that the benefits of a drug outweigh its risks." It covers an increasingly large array of medications.
But what does this grand plan require the drug companies to do?
Drug companies must create a database.
But for doctors who prescribe these antiarrhythmic medications and are board-certified to do so, we now have to perform a "one-time" re-certification that involves filling out a form and agreeing, in writing, to mandated patient appointment frequencies and minimum requirements for patient education that must be conducted during our office visits.
Such is the case with Pfizer's antiarrhythmic medication dofetilide (marketed as Tikosyn). Realize this "re-certification" comes AFTER we have all had to conduct a training regimen and were already registered with the company to prescribe the drug.
The REMS program, begun in 2008, grew more inclusive (and intrusive) after identification of a White House "crisis" involving prescription drug abuse of opioid analgesics that surfaced in April of this year. This edict has now trickled down to the clinical front lines of care with some very significant clinical consequences.
As clinical volumes rise, doctors are finding it increasingly difficult to reach the Utopian vision of frequent patient follow-up for drug surveillance for the pharmaceutical industry. Certainly, if there is clinical reason to do so (marginal renal function, higher-dose therapy, confounding medical issues) we see patients more frequently as needed. But in stable, relatively healthy patients who have a history of safely using these medications, we are left to wonder if the FDA's surveillance program has the potential to limit our ability to see new patients in favor of only managing established patients on chronic medication regimens that require close follow-up.
Clearly, there should be a balance. For many doctors (myself included) we have had to resort to using a nurse practitioner to assist with this requirement to offload the crush of such mandated patient visits. But for doctors in smaller, more rural settings where ancillary care providers are harder to come by, I suspect others will quickly saturate their clinics with regulated patient visits or else just not offer these medications to their patients.
This balance of safety and quality care to the oncoming tsunami of patients sure to hit our door in 2014 is an interesting dilemma not easily solved. Still, innovative ways to avoid top-down regulations that are crushing doctors with mandated (and often clinically unnecessary) care will go a long way to improving the quantity of care we are able to provide our growing population of patients.
-Wes
While post-market surveillance of medications is both necessary and warranted, it is interesting to me how the grunt work of this surveillance (and most other grand regulatory schemes) falls squarely on the backs of physicians rather than the drug companies who manufacture and profit from the medications.
Case in point: the FDA's REMS program. REMS stands for "Risk Evaluation and Mitigation Strategy" and is a program developed by the FDA "to manage known or potential serious risks associated with a drug product. It is required by the Food and Drug Administration (FDA) to ensure that the benefits of a drug outweigh its risks." It covers an increasingly large array of medications.
But what does this grand plan require the drug companies to do?
Drug companies must create a database.
But for doctors who prescribe these antiarrhythmic medications and are board-certified to do so, we now have to perform a "one-time" re-certification that involves filling out a form and agreeing, in writing, to mandated patient appointment frequencies and minimum requirements for patient education that must be conducted during our office visits.
Such is the case with Pfizer's antiarrhythmic medication dofetilide (marketed as Tikosyn). Realize this "re-certification" comes AFTER we have all had to conduct a training regimen and were already registered with the company to prescribe the drug.
The REMS program, begun in 2008, grew more inclusive (and intrusive) after identification of a White House "crisis" involving prescription drug abuse of opioid analgesics that surfaced in April of this year. This edict has now trickled down to the clinical front lines of care with some very significant clinical consequences.
As clinical volumes rise, doctors are finding it increasingly difficult to reach the Utopian vision of frequent patient follow-up for drug surveillance for the pharmaceutical industry. Certainly, if there is clinical reason to do so (marginal renal function, higher-dose therapy, confounding medical issues) we see patients more frequently as needed. But in stable, relatively healthy patients who have a history of safely using these medications, we are left to wonder if the FDA's surveillance program has the potential to limit our ability to see new patients in favor of only managing established patients on chronic medication regimens that require close follow-up.
Clearly, there should be a balance. For many doctors (myself included) we have had to resort to using a nurse practitioner to assist with this requirement to offload the crush of such mandated patient visits. But for doctors in smaller, more rural settings where ancillary care providers are harder to come by, I suspect others will quickly saturate their clinics with regulated patient visits or else just not offer these medications to their patients.
This balance of safety and quality care to the oncoming tsunami of patients sure to hit our door in 2014 is an interesting dilemma not easily solved. Still, innovative ways to avoid top-down regulations that are crushing doctors with mandated (and often clinically unnecessary) care will go a long way to improving the quantity of care we are able to provide our growing population of patients.
-Wes
Thursday, August 04, 2011
When Drugs Go Off the Radar
With the news that Pfizer is considering the possibility of making Lipitor (atorvastatin) over-the-counter (OTC), the FDA suddenly becomes "concerned:"
Mixed messages, anyone?
People only have so much money these days, so willful consumption of a relatively expensive OTC drug like Lipitor will likely only occur if (1) people feel they really need it and (2) if it proves to be affordable relative to the co-pay they have to pay from an insurer's plan. Pfizer realizes that most doctors consider their medication to be relatively safe. I suspect most cardiologists would agree with that sentiment.
Recently, doctors have seen the FDA overreach in clinical care. Their concerns about rhabdomyolysis with statins are important, but cardiologists like myself are baffled when the FDA makes random restrictions to the milligram dose of simvistatin when used in conjunction with other medications like Amiodarone: are there really any data supporting the use of limiting simvistatin use to 10 mg instead of 20 mg in their most recent recommendations? If so, where? Why did they change their earlier recommendation that 20mg was okay with Amiodarone? Or are they, instead, trying to minimize a side effect in the name of safety without data without balancing the possible incremental detriment to cardiovascular outcomes? I would venture that their recommendations like this that carry little clinical data subject doctors to greater individual liability risks compared to the greater risk of toxicity to their patients.
The FDA further notes that patients don't take Lipitor for symptoms, which is true. But I would venture that patients are probably smart enough to realize that if they start developing muscle aches after starting this drug, they are likely to stop the drug. Further, patients are paying for an increased percentage of their health care bill these days and that trend is likely to continue. Costs will drive compliance, and the best way to reduce costs is by market competition.
As far as liver toxicity with these drugs is concerned, the odds of seeing an elevated lipid panel on an asymptomatic patient on chronic Lipitor therapy is low. Certainly if it is found, stopping the drug might avert disaster. But the truth be known, when cardiologists see asymptomatic patients on Lipitor for their annual physicals, hepatic blood panels are drawn more for their defensive value against litigation than for their patients' clinical need. We justify this as "smart medicine" because we occassionally find an abnormality. But statistically, many "abnormal" lab tests are not acted upon clinically because "clinically" we know many of these "abnormals" are actually false positive abberations of the testing itself. In fact, when we do a panel of 10 chemistries from a single blood test, the odds that one of them will be abnormal can approach as much as 5%. Here's the truth: blood tests aren't perfect, either, so doctors practice clinical "judgement."
So I say, let Lipitor go over-the-counter. Give patients the benefit of the doubt regarding their intelligence and offer them an opportunity to drive down prices by having more options for their lipid management. Let them follow their own lipid and hepatic panels with home testing. I know it will be hard not to send all those electronic prescriptions via electronic medical records to all those pharmacy benefit managers in charge of all of those pharmacies out there, but maybe this is exactly the innovation our health care system needs to cut costs.
And who knows? Maybe our patients will actually do better for themselves than we give them credit for.
-Wes
But Pfizer likely faces an uphill battle because the U.S. Food and Drug Administration has previously rejected the idea of allowing over-the-counter versions of cholesterol drugs in the same class as Lipitor—known as statins—because of concerns that consumers aren't able to properly use the drugs without a doctor's guidance.Yes, this is the same FDA that has promoted direct-to-consumer advertising on TV and radio suddenly becoming "concerned" that patients might not make the right decisions about taking their medications.
FDA spokeswoman Shelly Burgess said prior research on proposed over-the-counter statins hasn't proven that most consumers will make correct decisions about taking the drugs. But she said the agency is open to discussing OTC statins, as long as companies are ready to demonstrate that consumers will make the right decisions.
Mixed messages, anyone?
People only have so much money these days, so willful consumption of a relatively expensive OTC drug like Lipitor will likely only occur if (1) people feel they really need it and (2) if it proves to be affordable relative to the co-pay they have to pay from an insurer's plan. Pfizer realizes that most doctors consider their medication to be relatively safe. I suspect most cardiologists would agree with that sentiment.
Recently, doctors have seen the FDA overreach in clinical care. Their concerns about rhabdomyolysis with statins are important, but cardiologists like myself are baffled when the FDA makes random restrictions to the milligram dose of simvistatin when used in conjunction with other medications like Amiodarone: are there really any data supporting the use of limiting simvistatin use to 10 mg instead of 20 mg in their most recent recommendations? If so, where? Why did they change their earlier recommendation that 20mg was okay with Amiodarone? Or are they, instead, trying to minimize a side effect in the name of safety without data without balancing the possible incremental detriment to cardiovascular outcomes? I would venture that their recommendations like this that carry little clinical data subject doctors to greater individual liability risks compared to the greater risk of toxicity to their patients.
The FDA further notes that patients don't take Lipitor for symptoms, which is true. But I would venture that patients are probably smart enough to realize that if they start developing muscle aches after starting this drug, they are likely to stop the drug. Further, patients are paying for an increased percentage of their health care bill these days and that trend is likely to continue. Costs will drive compliance, and the best way to reduce costs is by market competition.
As far as liver toxicity with these drugs is concerned, the odds of seeing an elevated lipid panel on an asymptomatic patient on chronic Lipitor therapy is low. Certainly if it is found, stopping the drug might avert disaster. But the truth be known, when cardiologists see asymptomatic patients on Lipitor for their annual physicals, hepatic blood panels are drawn more for their defensive value against litigation than for their patients' clinical need. We justify this as "smart medicine" because we occassionally find an abnormality. But statistically, many "abnormal" lab tests are not acted upon clinically because "clinically" we know many of these "abnormals" are actually false positive abberations of the testing itself. In fact, when we do a panel of 10 chemistries from a single blood test, the odds that one of them will be abnormal can approach as much as 5%. Here's the truth: blood tests aren't perfect, either, so doctors practice clinical "judgement."
So I say, let Lipitor go over-the-counter. Give patients the benefit of the doubt regarding their intelligence and offer them an opportunity to drive down prices by having more options for their lipid management. Let them follow their own lipid and hepatic panels with home testing. I know it will be hard not to send all those electronic prescriptions via electronic medical records to all those pharmacy benefit managers in charge of all of those pharmacies out there, but maybe this is exactly the innovation our health care system needs to cut costs.
And who knows? Maybe our patients will actually do better for themselves than we give them credit for.
-Wes
Wednesday, July 20, 2011
Our Polite New World of Rationing
technology (transcatheter aortic valve replacement [TAVR]), government,
industry and medicine will need to work in harmony.”
- David R. Holmes, Jr., MD, FACC
President, American College of Cardiology
Today, Edwards Lifesciences’ will request pre-market approval of its SAPIEN Transcatheter Heart Valve from the FDA's Circulatory Systems Devices Panel of the Medical Devices Advisory Committee. And for the first time, the groundwork for our complicated new era of health care rationing will be exposed.
To win an expensive technology on behalf of patients these days, there will have be "harmony" between doctors and their professional organizations and government regulators. If not, patients lose.
At issue is a transformative technology - another milestone forwarding medical innovation on behalf of some of our oldest and sickest patients: those with critical aortic stenosis who are too sick to undergo open heart surgery. Aortic stenosis tends to be a disease of the elderly that carries at least a 2-year 50% mortality when accompanied by a weakened heart muscle. Yet thanks to the wonders of careful engineering and some daring researchers that paired their expertise and lessons learned from a variety of disciples (cardiothoracic and peripheral vascular surgery, cardiology, and even cardiac electrophysiology), technigues and technology have combined to offer a percutaneous option for aortic valve replacement.
Everyone involved in this research (and even those who have watched from afar) knows this therapy works. Most believe in the long run, it will prove to be a safer option than open heart surgery in these patients.
But that's about where the harmony ends.
The new valve is expensive and so is the procedure to implant it. Although rumor, the valve itself might cost $20,000 US. Medicare (the insurer of the elderly) pays only 80% of the costs, typically, and has an arcane coding system that pays more for the code for aortic valve "replacement" than it does for aortic valve "insertion." (For goodness sakes, doctors, stop calling it TAVI and stick with TAVR, okay?!?) Will hospitals and insurers be able to afford a run on these devices? And what about Medicare that's already struggling with a huge unfunded liability?
And then there's the whole issue that doctors can't be trusted to do what's right for their patients anymore. They are uniformly greedy, at least in the eyes of the media and the regulators. They care about themselves more than their patients and thanks to a few unscrupulous doctors (and the fee-for-service system in which they work) ample evidence exists to contribute to this perception handsomely. Marcus Welby, MD: rest in peace.
But doctors still hold sway with their patients. For regulators, this is the biggest problem. Doctors, you see, get to stare directly into the eyes of the patients (and their families) as they discuss their principle problem: their narrowed aortic valve. We have to explain the options for treatment available: (1) doing nothing (and what will happen), (2) having open heart surgery (and what will happen), or (3)
Guess which option the patient is most likely to choose?
The fear with this new technology unleashed on the public, of course, is that the implant rate will reach a fever pitch as hospitals, ever hungry for the latest technology to tout, splash their cardiologists faces over billboards and national TV promoting
But if you really want to see all hell break loose, splash the images of a frail minority patient that was denied the option to receive a percutaneous valve on the basis of their age that turns to the media to expose their story.
Katie bar the door.
So we must be polite. We must demonstrate harmony. We must have databases. We must have panels of doctors and regulators and professional bodies assembled that sing Kum-By-Yah by their campfire is a great display of good will and uniform conviction to diffuse responsibility.
After all, rationing's a bitch.
-Wes
Friday, December 03, 2010
More on Biotronik's Exploding ('Venting'?) ICD
According to MedPageToday, it appears an earlier case report that was mysteriously withdrawn from the peer-reviewed journal Europace will soon be republished:
Several other issues:
A controversial article about problems with an implanted cardiac device -- published by and then withdrawn from the journal Europace -- has been resubmitted and is under review, according to the journal's editor.Hopefully, the journal will explain why they failed to notify their readers about the withdrawl as well. To withdraw an article of such signficance to their readership without explanation should not be tolerated by the scientific community.
"I expect that a decision on publication will be made very shortly," John Camm, MD, of St. George's University of London, told MedPage Today in an e-mail.
Several other issues:
- Perhaps even more concerning this whole ordeal has been the FDA's management of the device report made to them in May. It seems public reporting of Biotronik's filing did not appear on the MAUDE database until after my blog post was published in October.
If this is their policy to withhold reports in patients that are injured for this length of time irrespective of "cause," there are bigger concerns with the government's policies that should be immediately addressed. - I should also explain my rationale for my "defensive blogging" earlier, too.
The Fair Use Act of US Copyright law has been a favorite place for malicious lawyers to attack bloggers who republish content in their blogs. Irrespective of whether or not one could defend their actions in the court of law on the consitutional basis of "free speech."
But when challenged, as soon as a lawyer gets involved, thanks to the large costs involved, you've "lost" your case even before going to trial. I did not need that expense at the time, so I caved and withdrew the pictures I had published (note: they have since been republished on the blog Cardiobrief.org, courtesy of Google cache, but have included a pdf of my copy of the entire case report in this blog post).
Friday, November 12, 2010
FDA Proposes New Cigarette Labels
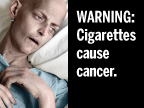 The FDA will soon require new cigarette package labeling to deter smoking. So in politically-correct governmental fashion, they are asking which labels you'd like to see. (You can pick your favorites here.) My personal favorite (so far) is the one shown to the left, but its impact factor pales in comparison to this example found in England. (That, my friends, is cancer!)
The FDA will soon require new cigarette package labeling to deter smoking. So in politically-correct governmental fashion, they are asking which labels you'd like to see. (You can pick your favorites here.) My personal favorite (so far) is the one shown to the left, but its impact factor pales in comparison to this example found in England. (That, my friends, is cancer!) Ironically, it appears the FDA isn't too sure how forceful it should be in these warnings about the dangers of smoking. They offer a cornucopia of milquetoast labeling options - many of which contain cartoons. Might such unrealistic portrayals defy they hard-hitting message they want to project? Worse, at least one cartoon (seen here) even seems to promote cigarettes AND drug use together!
Ironically, it appears the FDA isn't too sure how forceful it should be in these warnings about the dangers of smoking. They offer a cornucopia of milquetoast labeling options - many of which contain cartoons. Might such unrealistic portrayals defy they hard-hitting message they want to project? Worse, at least one cartoon (seen here) even seems to promote cigarettes AND drug use together!  In an even more astonishing example, some images almost make me what to take up smoking so I can blow big bubbles. Since I could never do this well before, maybe I should take up smoking! Seriously, is an empowerment message what the government wants to portray?
In an even more astonishing example, some images almost make me what to take up smoking so I can blow big bubbles. Since I could never do this well before, maybe I should take up smoking! Seriously, is an empowerment message what the government wants to portray?Make these labels big, ugly, and real.
Anything else is a waste of taxpayer's money.
-Wes
Tuesday, October 19, 2010
Finally: Dabigatran - A New Oral Anticoagulant is Approved by the FDA
A new era of non-valvular atrial fibrillation management has arrived.
Today, the FDA approved the first new anticoagulant in fifty years, dabigatran (marketed by Boehringer Ingelheim Pharmaceuticals Inc. under the trade name Pradaxa®) for stroke prevention in patients with non-valvular atrial fibrillation. The move was widely anticipated after the drug's unanimous 9-0 FDA advisory panel recommendation for approval a month ago. The drug will be available in 75 an 150 milligram dosages and is taken twice daily.
Patients at high risk of thromboembolism from non-valvular atrial fibrillation are candidates for the drug including patients with previous stroke or transient ischemic attack, a left ventricular ejection fraction of less than 40%, New York Heart Association class II or higher heart-failure symptoms within 6 months before screening for the medication, and an age of at least 75 years or an age of 65 to 74 years plus diabetes mellitus, hypertension, or coronary artery disease.
Patients who should NOT receive the drug include patients with the presence of a severe heart-valve disorder, stroke within 14 days or severe stroke within the last 6 months, a condition that increases the risk of hemorrhage, a creatinine clearance of less than 30 ml per minute, active liver disease, or pregnancy. Patients taking quinidine should also not take the medication because of a significant drug interaction.
The drug does not typically require measurement of blood thinning levels (prothrombin times expressed as and international normalized ratio (INR) of clotting time to a standard clotting control).
The approval was based on the prospective, randomized RE-LY trial recently published in the New England Journal of Medicine that compared the safety and efficancy of two doses of dabigatran (110 mg and 150 mg twice daily) to conventional warfarin (Coumadin®) therapy in 18,113 patients:
The drug's most common side effect was dyspepsia (GI upset) but liver enzyme elevations were not any different than that seen with warfarin.
The questions now are two: (1) when will it be available and (2) how much will it cost?
Typically it takes about 3 to 6 months to finalize product packaging, labeling and distribution after a drug is approved (others may have info they can share here). As far as price - my bet is that it's going to cost about ten times that of warfarin - I'd estimate $6 to $9 per day (another author suggested the anticipated cost of dabigatran in the United States, as calculated on the basis of its cost in Canada, would be approximately $7,000 to $9,000 per patient-year (four to five times the cost of warfarin, despite the increased physician and laboratory costs required to monitor the international normalized ratio [INR])). One researcher from the RE-LY trial countered:
-Wes
Addendum: MedPageToday: Dabigatran: The Case of the Missing 110-mg Dose
Today, the FDA approved the first new anticoagulant in fifty years, dabigatran (marketed by Boehringer Ingelheim Pharmaceuticals Inc. under the trade name Pradaxa®) for stroke prevention in patients with non-valvular atrial fibrillation. The move was widely anticipated after the drug's unanimous 9-0 FDA advisory panel recommendation for approval a month ago. The drug will be available in 75 an 150 milligram dosages and is taken twice daily.
Patients at high risk of thromboembolism from non-valvular atrial fibrillation are candidates for the drug including patients with previous stroke or transient ischemic attack, a left ventricular ejection fraction of less than 40%, New York Heart Association class II or higher heart-failure symptoms within 6 months before screening for the medication, and an age of at least 75 years or an age of 65 to 74 years plus diabetes mellitus, hypertension, or coronary artery disease.
Patients who should NOT receive the drug include patients with the presence of a severe heart-valve disorder, stroke within 14 days or severe stroke within the last 6 months, a condition that increases the risk of hemorrhage, a creatinine clearance of less than 30 ml per minute, active liver disease, or pregnancy. Patients taking quinidine should also not take the medication because of a significant drug interaction.
The drug does not typically require measurement of blood thinning levels (prothrombin times expressed as and international normalized ratio (INR) of clotting time to a standard clotting control).
The approval was based on the prospective, randomized RE-LY trial recently published in the New England Journal of Medicine that compared the safety and efficancy of two doses of dabigatran (110 mg and 150 mg twice daily) to conventional warfarin (Coumadin®) therapy in 18,113 patients:
Rates of the primary outcome (stroke and systemic embolization) were 1.69% per year in the warfarin group, as compared with 1.53% per year in the group that received 110 mg of dabigatran (relative risk with dabigatran, 0.91; 95% confidence interval [CI], 0.74 to 1.11; P < 0.001 for noninferiority) and 1.11% per year in the group that received 150 mg of dabigatran (relative risk, 0.66; 95% CI, 0.53 to 0.82; P < 0.001 for superiority). The rate of major bleeding was 3.36% per year in the warfarin group, as compared with 2.71% per year in the group receiving 110 mg of dabigatran (P=0.003) and 3.11% per year in the group receiving 150 mg of dabigatran (P=0.31). The rate of hemorrhagic stroke was 0.38% per year in the warfarin group, as compared with 0.12% per year with 110 mg of dabigatran (P < 0.001) and 0.10% per year with 150 mg of dabigatran (P < 0.001). The mortality rate was 4.13% per year in the warfarin group, as compared with 3.75% per year with 110 mg of dabigatran (P=0.13) and 3.64% per year with 150 mg of dabigatran (P=0.051).It should be noted that the FDA did not approve the lower 110 mg dose of the medication.
The drug's most common side effect was dyspepsia (GI upset) but liver enzyme elevations were not any different than that seen with warfarin.
The questions now are two: (1) when will it be available and (2) how much will it cost?
Typically it takes about 3 to 6 months to finalize product packaging, labeling and distribution after a drug is approved (others may have info they can share here). As far as price - my bet is that it's going to cost about ten times that of warfarin - I'd estimate $6 to $9 per day (another author suggested the anticipated cost of dabigatran in the United States, as calculated on the basis of its cost in Canada, would be approximately $7,000 to $9,000 per patient-year (four to five times the cost of warfarin, despite the increased physician and laboratory costs required to monitor the international normalized ratio [INR])). One researcher from the RE-LY trial countered:
It should also be kept in mind that total direct andGiven its cost, I suspect it will be hard for insurers to swallow this drug at first and coverage may not be immediately available, but hopefully the superior convenience and stroke prevention will justify the drug's initial price. Fortunately, other thrombin inhibitors will soon arrive to offer price competition to dabigatran's exclusive first-to-market reign.
indirect costs for management of anticoagulation with warfarin far exceed the cost of the drug. In a recent study, the direct costs during the first year of nticoagulation with warfarin in primary care were calculated at Swedish krona 16,244, corresponding to U.S. $2,230. This does not include expenses to patients for travel to the laboratory, lost time from work, or an accompanying caregiver.
-Wes
Addendum: MedPageToday: Dabigatran: The Case of the Missing 110-mg Dose
Saturday, October 09, 2010
Biotronik Responds to Europace Exploding ICD Case Report Article Withdrawl
Yesterday evening, I met with Rex Richmond (Vice President of Marketing) and Dan Schlewitz (Executive Vice President U.S.A. Sales) from Biotronik, Inc. to hear their side of the controversy surrounding the withdrawal of a case report previously published in Europace pertaining to an "exploding" Biotronik implantable cardiac defibrillator (ICD). They claimed they have been transparent regarding the reporting of this incident to the FDA, filing their incident report with the FDA within seven days of the event (within thirty days is required). Mr. Richmond granted me permission to release their internal company memo (pdf) circulated to their employees regarding the case report.
I must supply several comments.
First, regarding why the case report was withdrawn, they state in their memo:
Second is the issue of FDA reporting mentioned in the memo:
-Wes
Reference: pdf of my personal copy of withdrawn Europace case report obtained online 5 Oct 2010 before its withdrawl with timeline of the article's submission/revision dates.
CORRECTION - 12:55pm CST 12 Oct 2010: Here's a link to the FDA report regarding this incident filed on the FDA's MAUDE database. I apologize for the inaccuracy.
Addendum 09:45 AM CST 12 Oct 2010 - Reactions from around the web:
Cardiobrief: "The Plot Thickens in the Case of the Exploding ICD"
Happy Hospitalist: "Exploding ICD Gets Biotronik Response"
I must supply several comments.
First, regarding why the case report was withdrawn, they state in their memo:
The author reported that after submitting the case report to Europace in June, further analysis was conducted but not included in the original report. As such, there are inaccuracies that need to be corrected. Specifically, the author stated that the term "explosion" was not accurate given that the device was distorted, but had not exploded as previously described. The author also observed that while this is the first such incident with a BIOTRONIK device, it is not in fact the first experience of a battery overheating in the industry.Note that the updated MDA was submitted to the FDA 6 June 2010. The published article was submitted to Europace 29 June 2010 and accepted after revision 16 August 2010. Wouldn't these "inaccuracies" have been corrected with the revisions submitted back to Europace following their review well before the 27 Sep 2010 publishing date?
Second is the issue of FDA reporting mentioned in the memo:
Where do reports like this end up? Aren’t they public?I do not know why the FDA would not have published the MDA's received in May with revisions in June on their website by 31 August 2010 (August data appear to have the latest updates available online) if the original MDA was submitted in May as Biotronik suggests. Does it really take this long to review MDA's before they are published? It is one thing to verify an MDA from the public before publishing on the FDA MAUDE database, but a manufacturer's MDA regarding their own device should be published without delay.
Yes, they do become public. The FDA posts MDR’s to their online Manufacturer and User Facility Device Experience (MAUDE) database at the following address:
http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfMAUDE/TextSearch.cfm
(ed: Emphasis mine) Although the FDA has our MDR, it has yet to be posted to the database.
-Wes
Reference: pdf of my personal copy of withdrawn Europace case report obtained online 5 Oct 2010 before its withdrawl with timeline of the article's submission/revision dates.
CORRECTION - 12:55pm CST 12 Oct 2010: Here's a link to the FDA report regarding this incident filed on the FDA's MAUDE database. I apologize for the inaccuracy.
Addendum 09:45 AM CST 12 Oct 2010 - Reactions from around the web:
Cardiobrief: "The Plot Thickens in the Case of the Exploding ICD"
Happy Hospitalist: "Exploding ICD Gets Biotronik Response"
Monday, September 13, 2010
Midodrine's Updates
On 16 August 2010, the FDA published a "Proposal to Withdraw Low Blood Pressure Drug" about the generic blood pressure-raising medication, midodrine:
Two days later on 18 August 2010, Shire pharmaceuticals announced they will stop manufacturing midodrine 30 September 2010, adding fuel to the patient backlash of the FDA's prior announcement:
First, once the FDA approves a drug with strings attached, they have an obligation to assure to the public that those strings mean something long before a drug goes generic. Why impose regulatory requirements if they're not going to enforce those requirements in a timely fashion? We are left to wonder how many patients might have been harmed by their inaction had there been a problem.
Secondly, news agencies now appear to be our best source of new information about updates on drugs and their availability. This should be of considerable concern to physicians who need to verify the legitimacy of information published in the mainstream media. Admittedly, the New York Times got the story right, but such is not always the case with media reports about drugs and innovations in medicine. At the very least, the FDA should provide midodrine updates at the same time this information is provided to the media. Verifiable information is increasingly needed in our new era of web-enabled patients and doctors.
Third, this was one small example of the power of government to intervene on our patients' care rapidly and without warning to the populace at large. Fortunately, the public outcry over their action was not ignored, but patients should be aware that litigation against the government should harm arise as a result of such an action would be very difficult and expensive to prosecute.
Hopefully, the FDA has learned a few lessons with this fiasco.
One thing's for sure: you can bet our patients did.
-Wes
The U.S. Food and Drug Administration today proposed to withdraw approval of the drug midodrine hydrochloride, used to treat the low blood pressure condition orthostatic hypotension, because required post-approval studies that verify the clinical benefit of the drug have not been done.Phone lines were flooded. Doctors scratched their heads because the "options" for alternate therapies to replace this drug amounted to exactly... none.
Patients who currently take this medication should not stop taking it and should consult their health care professional about other treatment options.
Two days later on 18 August 2010, Shire pharmaceuticals announced they will stop manufacturing midodrine 30 September 2010, adding fuel to the patient backlash of the FDA's prior announcement:
Shire plc (LSE: SHP, NASDAQ: SHPGY) acquired ProAmatine (ed's note: trade name for midodrine) as a part of the acquisition of Roberts Pharma in 1999 and Shire conducted and completed the post marketing trials that the FDA required. The FDA, however, viewed these trials as inconclusive and required that additional trials be conducted for ProAmatine to maintain its marketing authorization. As a result, Shire elected to withdraw the product effective September 30, 2010 and notified the FDA in November 2009 and healthcare professionals earlier this year of this decision. Shire's withdrawal of the NDA was not related to any concerns regarding the safety of ProAmatine.Doctors scrambled. Patients complained. Finally, news about the FDA recanting came via a New York Times article on 3 September 2010:
The agency’s flip-flop demonstrates the difficult choices regulators face in policing the nation’s drug market. Cracking down on drug makers sometimes means stranding desperate patients. And now that Congress has given the Food and Drug Administration greater powers to insist on better information about life-saving medicines, such disputes may become more common.Sadly, not until a 10 September 2010 press release from the FDA could the information published in the New York Times be confirmed and a plan of action taken to assure patients and their physicians aren't blind-sided by the FDA's actions on this matter:
Since the agency notified the manufacturers of the proposed withdrawal of midodrine, we have heard from professional organizations, doctors, and patients about their concern about losing access to midodrine. We have also heard from organizations that have expressed interest in conducting the clinical studies necessary to establish effectiveness, as well as doctors and patients who support the conduct of such studies. FDA intends to work with Shire, the generic manufacturers, and other organizations to discuss the data that are necessary to establish the efficacy of midodrine. FDA will post this information in a public docket.So what have we learned with this regulatory fiasco?
First, once the FDA approves a drug with strings attached, they have an obligation to assure to the public that those strings mean something long before a drug goes generic. Why impose regulatory requirements if they're not going to enforce those requirements in a timely fashion? We are left to wonder how many patients might have been harmed by their inaction had there been a problem.
Secondly, news agencies now appear to be our best source of new information about updates on drugs and their availability. This should be of considerable concern to physicians who need to verify the legitimacy of information published in the mainstream media. Admittedly, the New York Times got the story right, but such is not always the case with media reports about drugs and innovations in medicine. At the very least, the FDA should provide midodrine updates at the same time this information is provided to the media. Verifiable information is increasingly needed in our new era of web-enabled patients and doctors.
Third, this was one small example of the power of government to intervene on our patients' care rapidly and without warning to the populace at large. Fortunately, the public outcry over their action was not ignored, but patients should be aware that litigation against the government should harm arise as a result of such an action would be very difficult and expensive to prosecute.
Hopefully, the FDA has learned a few lessons with this fiasco.
One thing's for sure: you can bet our patients did.
-Wes
Subscribe to:
Comments (Atom)